High-throughput computation of important properties of chemical systems with the latest theoretical chemistry methods is a fundamental quest from the realm of computational chemistry. We have been working on implementing our theory on existing or new quantum molecular dynamics computer programs to fulfill the requirement. We hope these programs can build a bridge between theorists and experimentalists, making convenient use of our novel path integral molecular dynamics and nonadiabatic dynamics methods.
In the current programs, PSiQMD serves as an overall platform collecting our theoretical and coding process in (path-integral) molecular dynamics and nonadiabatic dynamics. PSiNaD is a nonadiabatic dynamics-only branch of PSiQMD, while hNaF is specified for model dynamics and method developments with the support of ultra-high throughput heterogeneous hardwares. A list of our programs is as follows:
Currently we have been working on adding the following features to the corresponding existing programs:
PSiNaD (Phase Space Integrated Nonadiabatic Dynamics) is an open-source framework specifically designed for simulating chemical and physical dynamics at atomic and molecular scales in condensed matter. It excels in classical and quantum dynamics simulations, accommodating small systems, few-body systems, reduced systems, and large many-particle systems such as molecules and condensed matter. PSiNaD offers a robust, versatile platform with multiple interfaces for developing advanced algorithms, ensuring ease of use and accessibility with third-party language wrappers, such as Python.
PSiNaD features a comprehensive array of models and solvers, including harmonic models, forcefield models, and ab initio interfaces, meeting diverse simulation needs. It supports a wide range of computational approaches, from classical dynamics based on Newtonian and Hamiltonian mechanics to sophisticated quantum dynamics incorporating quantum phase space approximations, quantum trajectories, path integral techniques, influence functional methods, and semi-classical wavepackets.
Continual development efforts focus on expanding the library of models and solvers, with community contributions highly encouraged. To further extend its accessibility, PSiNaD is now available as a Python package (PyPSiNaD).
Related papers:
Xin He, Haocheng Lu, Jian Liu*, "PSiNaD: An Extensible Solution for Nonadiabatic Dynamics Based on Phase Space Formulations" (in preparation)
Baihua Wu, Bingqi Li, Xin He, Xiangsong Cheng, Jiajun Ren, Jian Liu*, "Nonadiabatic Field: A Conceptually Novel Approach for Nonadiabatic Quantum Molecular Dynamics", Journal of Chemical Theory and Computation, 21(8), 3775-3813 (2025) [https://doi.org/10.1021/acs.jctc.5c00181]
PSiQMD includes molecular dynamics, path integral molecular dynamics, path integral Liouville dynamics, nonadiabatic molecular dynamics, and so on. It not only collects progress of our coding in
and for
but also includes phase space quantum dynamics approaches and other trajectory-based dynamics/thermodynamics methods for real molecular systems.
Related papers:
Xin He, Haocheng Lu, Jian Liu*, "PSiNaD: An Extensible Solution for Nonadiabatic Dynamics Based on Phase Space Formulations" (in preparation)
Weihao Liang, Sihan Wang, Cong Wang, Weizhou Wang, Xinchen She, Chongbin Wang, Jiushu Shao, Jian Liu*, "An Efficient Integrator Scheme for Sampling the (Quantum) Isobaric-Isothermal Ensemble in (Path Integral) Molecular Dynamics", Journal of Chemical Theory and Computation, 21, 13, 6394–6409 (2025) [https://doi.org/10.1021/acs.jctc.5c00573]
Baihua Wu, Bingqi Li, Xin He, Xiangsong Cheng, Jiajun Ren, Jian Liu*, "Nonadiabatic Field: A Conceptually Novel Approach for Nonadiabatic Quantum Molecular Dynamics", Journal of Chemical Theory and Computation, 21(8), 3775-3813 (2025) [https://doi.org/10.1021/acs.jctc.5c00181]
Zhijun Zhang, Xinzijian Liu, Kangyu Yan, Mark Tuckerman, Jian Liu*, "A unified efficient thermostat scheme for the canonical ensemble with holonomic or isokinetic constraints via molecular dynamics", The Journal of Physical Chemistry A, 123, 28, 6056-6079 (2019). [https://doi.org/10.1021/acs.jpca.9b02771][pdf version]
Zhijun Zhang, Xinzijian Liu, Zifei Chen, Haifeng Zheng, Kangyu Yan, Jian Liu*, "A unified thermostat scheme for efficient configurational sampling for classical/quantum canonical ensembles via molecular dynamics", The Journal of Chemical Physics, 147, 034109 (2017) [http://dx.doi.org/10.1063/1.4991621] [Supplementary material] [arXiv preprint] [pdf version]
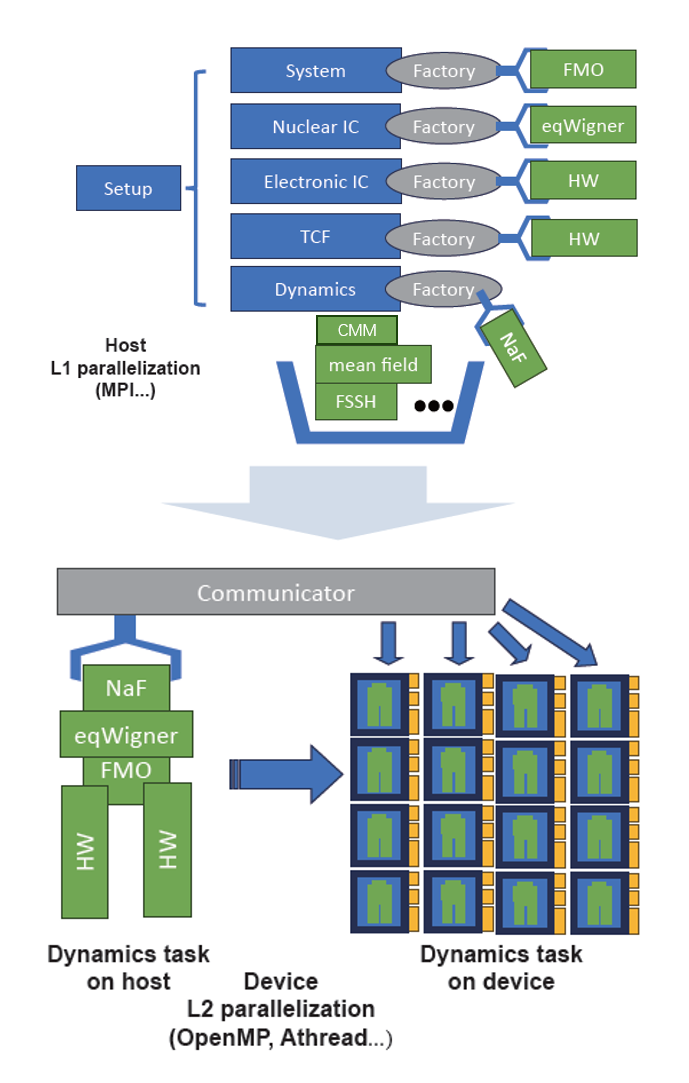
hNaF is a nonadiabatic dynamics program designed for ultra-high throughput model tests and method developments on heterogeneous computation platforms. Homemade computational platforms are usually equipped with specially designed heterogeneous hardwares, parallel interfaces and limited supports to the programming languages. By modularizing the computation tasks and separating out the parallel layers, hNaF enables us to test our new nonadiabatic dynamics methods rapidly, making full use of the homemade hardwares, for example, the new Sunway supercomputer.
hNaF can be viewed and cloned from the following repository: jianliugroup/hNaF
Related papers:
Baihua Wu, Bingqi Li, Xin He, Xiangsong Cheng, Jiajun Ren, Jian Liu*, "Nonadiabatic Field: A Conceptually Novel Approach for Nonadiabatic Quantum Molecular Dynamics", Journal of Chemical Theory and Computation, 21(8), 3775-3813 (2025) [https://doi.org/10.1021/acs.jctc.5c00181]
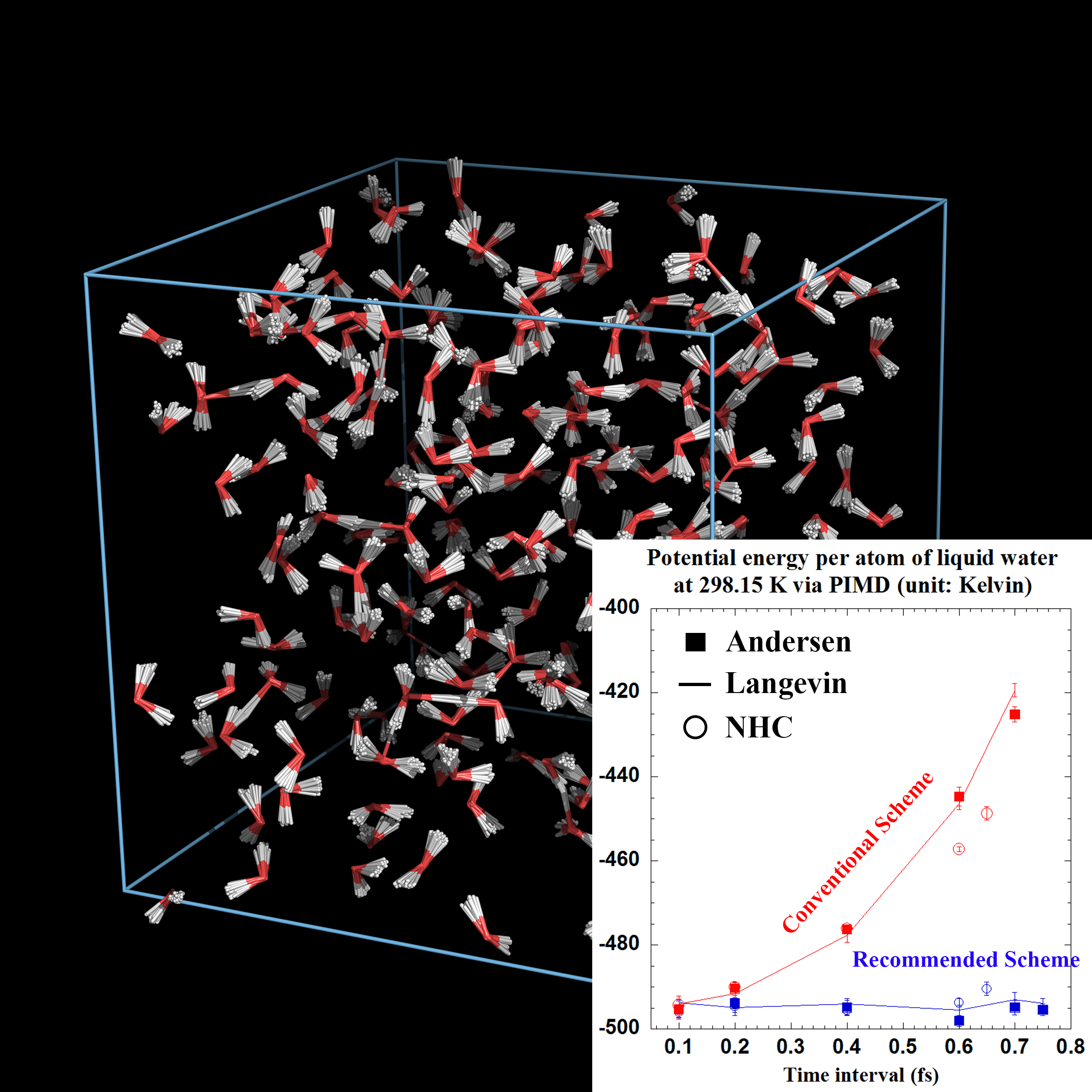

[NEW!] We have implemented a recommended "middle" scheme for the isobaric-isothermal ensemble in molecular dynamics packages, e.g., AMBER, GROMACS, and DL-POLY. An detailed description is on the way. The implementations can be viewed and cloned from our github repository,
We have implemented the efficient "middle" thermostat scheme into the lastest version of AmberTools since 2019. It can be obtained by following the instructions on the official website. For the users who can only use AmberTools18, the updated files for the "middle" scheme for AmberTools18 can be downloaded from the GitHub webpage.
The tutorial for the "middle" scheme is available on the webpage. One is also encouraged to read the AMBER manual for more details. The "middle" thermostat scheme has been implemented for MD for classical statistics as well as primitive PIMD for quantum statistics. It can be used together with QM/MM and enhanced sampling techniques (such as replica exchange) in AMBER. One can do ab initio PIMD or MD for the canonical ensemble by using AMBER combined with other quantum chemistry packages.
Related papers:
David A. Case, David S. Cerutti, Vinícius Wilian D. Cruzeiro, Thomas A. Darden, Robert E. Duke, Mahdieh Ghazimirsaeed, George M. Giambaşu, Timothy J. Giese, Andreas W. Götz, Julie A. Harris, Koushik Kasavajhala, Tai-Sung Lee, Zhen Li, Charles Lin, Jian Liu, Yinglong Miao, Romelia Salomon-Ferrrer, Jana Shen, Ryan Snyder, Jason Swails, Ross C. Walker, Jinan Wang, Xiongwu Wu, Jinzhe Zeng, Thomas E. Cheatham III, Daniel R. Roe, Adrian Roitberg, Carlos Simmerling, Darrin M. York, Maria C. Nagan*, Kenneth M. Merz Jr.*, "Recent Developments in Amber Biomolecular Simulations", Journal of Chemical Information and Modelling, 65, 15, 7835-7843 (2025).
Weihao Liang, Sihan Wang, Cong Wang, Weizhou Wang, Xinchen She, Chongbin Wang, Jiushu Shao, Jian Liu*, "An Efficient Integrator Scheme for Sampling the (Quantum) Isobaric-Isothermal Ensemble in (Path Integral) Molecular Dynamics Simulations", Journal of Chemical Theory and Computation, 21, 13, 6394–6409 (2025) [https://doi.org/10.1021/acs.jctc.5c00573]
David A. Case, Hasan Metin Aktulga, Kellon Belfon, David S. Cerutti, G. Andrés Cisneros, Vinícius Wilian D. Cruzeiro, Negin Forouzesh, Timothy J. Giese, Andreas W. Götz, Holger Gohlke, Saeed Izadi, Koushik Kasavajhala, Mehmet C. Kaymak, Edward King, Tom Kurtzman, Tai-Sung Lee, Pengfei Li, Jian Liu, Tyler Luchko, Ray Luo, Madushanka Manathunga, Matias R. Machado, Hai Minh Nguyen, Kurt A. O’Hearn, Alexey V. Onufriev, Feng Pan, Sergio Pantano, Ruxi Qi, Ali Rahnamoun, Ali Risheh, Stephan Schott-Verdugo, Akhil Shajan, Jason Swails, Junmei Wang, Haixin Wei, Xiongwu Wu, Yongxian Wu, Shi Zhang, Shiji Zhao, Qiang Zhu, Thomas E. Cheatham III, Daniel R. Roe, Adrian Roitberg, Carlos Simmerling, Darrin M. York, Maria C. Nagan, Kenneth M. Merz Jr. , "AmberTools", Journal of Chemical Information and Modeling, 63(20), 6183-6191 (2023) [https://pubs.acs.org/doi/10.1021/acs.jcim.3c01153] [pdf version]
Zhaoxi Sun, Payam Kalhor, Yang Xu, Jian Liu*,"Extensive Numerical Tests of the Leapfrog Integrator in the Middle Thermostat Scheme in Molecular Simulations", Chinese Journal of Chemical Physics, 34, 6, 932-948 (2021) [https://doi.org/10.1063/1674-0068/cjcp2111242]
Zhijun Zhang, Xinzijian Liu, Kangyu Yan, Mark Tuckerman, Jian Liu*, "A unified efficient thermostat scheme for the canonical ensemble with holonomic or isokinetic constraints via molecular dynamics", The Journal of Physical Chemistry A, 123, 28, 6056-6079 (2019). [https://doi.org/10.1021/acs.jpca.9b02771][pdf version]
Xinzijian Liu, Jian Liu*, "Path integral molecular dynamics for exact quantum statistics of multi-electronic-state systems", The Journal of Chemical Physics, 148, 102319 (2018) [https://doi.org/10.1063/1.5005059] [arXiv preprint] [pdf version]
Zhijun Zhang, Kangyu Yan, Xinzijian Liu, Jian Liu*, "A leap-frog algorithm-based efficient unified thermostat scheme via molecular dynamics", Chinese Science Bulletin, 63(33), 3467-3483(2018) [http://engine.scichina.com/doi/10.1360/N972018-00908 ] [pdf version]
Dezhang Li, Zifei Chen, Zhijun Zhang, Jian Liu*, "Understanding molecular dynamics with stochastic processes via real or virtual dynamics", Chinese Journal of Chemical Physics, 30, 735 (2017) [http://dx.doi.org/10.1063/1674-0068/30/cjcp1711223] [pdf version from the CJCP website] [arXiv preprint] [pdf version]
Zhijun Zhang, Xinzijian Liu, Zifei Chen, Haifeng Zheng, Kangyu Yan, Jian Liu*, "A unified thermostat scheme for efficient configurational sampling for classical/quantum canonical ensembles via molecular dynamics", The Journal of Chemical Physics, 147, 034109 (2017) [http://dx.doi.org/10.1063/1.4991621] [Supplementary material] [arXiv preprint] [pdf version]
Jian Liu*, Dezhang Li, Xinzijian Liu, "A simple and accurate algorithm for path integral molecular dynamics", The Journal of Chemical Physics, 145, 024103 (2016) [http://dx.doi.org/10.1063/1.4954990] [Supplementary material] [arXiv preprint (Paper + Supplemental materical)] [pdf version]
Jian Liu*, Zhijun Zhang, "Path integral Liouville dynamics: Applications to infrared spectra of OH, water, ammonia, and methane", The Journal of Chemical Physics, 144, 034307 (2016) [http://dx.doi.org/10.1063/1.4939953] [pdf version]